Clinical Pharmacology Stendra
|
CLINICAL PHARMACOLOGY
Mechanism of Action
The physiologic mechanism of erection of the penis involves release of nitric oxide (NO) in the corpus cavernosum during sexual stimulation. NO then activates the enzyme guanylate cyclase, which ends in increased levels of cGMP, producing smooth muscle relaxation in the corpus cavernosum and allowing inflow of blood. Avanafil doesn't have direct relaxant effect on isolated human corpus cavernosum, but enhances the issue of NO by inhibiting PDE5, and that is to blame for degradation of cGMP in the corpus cavernosum. Because sexual stimulation has to initiate any local discharge of nitric oxide supplements, the inhibition of PDE5 doesn't have effect without sexual stimulation.
Studies in vitro indicate that avanafil is selective for PDE5. Its effect might be more potent on PDE5 than you are on other known phosphodiesterases (higher than 100-fold for PDE6; greater than 1,000-fold for PDE4, PDE8 and PDE10; over 5,000-fold for PDE2 and PDE7; over 10,000-fold for PDE1, PDE3, PDE9, and PDE11). Avanafil is higher than 100-fold less assailable for PDE5 than PDE6, which is found in the retina and it's the cause of phototransduction. In combination with human corpus cavernosum smooth muscle, PDE5 can be within other tissues including platelets, vascular and visceral involuntary muscle, and striated muscle, brain, heart, liver, kidney, lung, pancreas, prostate, bladder, testis, and seminal vesicle. The inhibition of PDE5 of these tissues by avanafil is a basis for the enhanced platelet anti-aggregatory activity of NO seen in vitro and peripheral vasodilatation in vivo.
Pharmacodynamics
Results of STENDRA on Erectile Response
A single-blind, placebo-controlled, single-dose trial of 82 patients with either organic and/or psychogenic ED, visual sexual stimulation resulted in improved erections after STENDRA administration in comparison to placebo, as assessed by a goal measurement of hardness and duration of erections (RigiScan®). Efficacy was assessed by RigiScan at discrete time intervals including 20 – 40 minutes after dosing to 100 – 2 hours after dosing.
Results of STENDRA on Blood Pressure
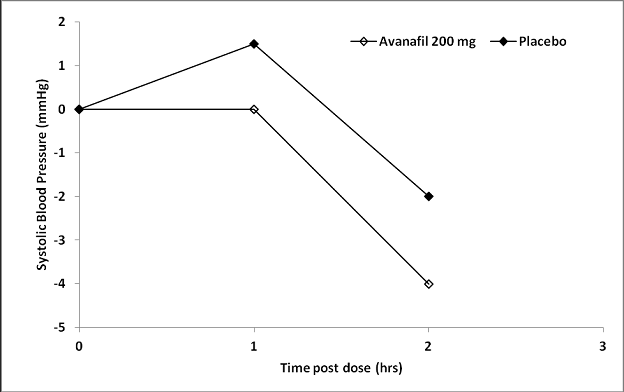
Single oral doses of STENDRA (200 mg) administered to
healthy male volunteers triggered mean changes from baseline in
systolic/diastolic high blood pressure of -5.3/-3.7 mmHg at 60 minutes after dosing,
in comparison with mean changes from baseline in the placebo gang of 2.7/-
Figure 1: Median Differ from Baseline in Sitting Systolic High blood pressure, Healthy Volunteers Day 4
Effects on Cardiac Electrophysiology
The effect of single 100 or 800 mg doses of STENDRA on the QT interval were evaluated inside a randomized, double-blind, placebo, and active (moxifloxacin) –controlled crossover study in 52 healthy male subjects aged 18 to 45 years. There initially were no significant effects on the 100 mg dose. The mean QTc (Fridericia QT correction) for avanafil 800 mg, in accordance with placebo was 9.4 milliseconds (two-sided 90% CI=7.2, 11.6). An 800 mg dose of STENDRA (4x the very best recommended dose) was chosen as this dose yields exposures higher than those observed upon co-administration of avanafil with strong CYP3A4 inhibitors. A double-blind, randomized, placebo- and active-controlled (moxifloxacin), thorough QT/QTc trial of STENDRA (100 and 800 mg) in healthy male subjects indicated that STENDRA did not cause any significant adjustments to QTc interval or ventricular repolarization.
Effects of STENDRA on Blood Pressure When Administered with Nitrates
In a very clinical pharmacology trial, one particular dose of STENDRA 200 mg was proven to potentiate the hypotensive effect of nitrates. The use of STENDRA in patients taking any type of nitrates is contraindicated [see Contraindications].
An effort was conducted to evaluate the amount of interaction between nitroglycerin and STENDRA, should nitroglycerin be required to pull up quickly situation after STENDRA was taken. It was just one-center, double-blind, randomized, 3-way crossover trial of healthy males from 30 to 60 years of age. Subjects were divided among 5 trial groups using the trial group being based upon the time interval between treatment with trial drug and glyceryl trinitrate administration. Subjects were assigned to trial groups sequentially and hemodynamic results from the previous group were reviewed for serious adverse events (SAEs) ahead of the next group received treatment. Each subject was dosed with all 3 study drugs (STENDRA 200 mg, sildenafil citrate 100 mg, and placebo) in random order. Subjects were administered a single dose of 0.4 mg sublingual nitroglycerin (NTG) at pre-specified time points, following their dose of trial drug (0.5, 1, 4, 8 or 12 hours). Overall, 14 (15%) subjects treated with placebo and 28 (28%) subjects treated with avanafil, had clinically significant decreases in standing SBP, understood to be more than or corresponding to 30 mmHg lowering in SBP, after glyceryl trinitrate administration. Mean maximum decreases are shown in Table 4.
Table 4: Mean Maximum Decreases from Baseline in Sitting and Standing Systolic High blood pressure/Diastolic Blood Pressure (mmHg) following Placebo or 200 mg STENDRA with 0.4 mg sublingual nitroglycerin
|
Placebo with nitroglycerin Sitting Standing |
13.4/11.8 21.1/16.5 |
|
STENDRA with nitroglycerin Sitting Standing |
21.6/18.2 28.0/23.5 |
Like other PDE5 inhibitors, STENDRA administration with nitrates is contraindicated. Within a patient who's taken STENDRA, where nitrate administration is deemed medically necessary in the life threatening situation, no less than 12 hours should elapse following last dose of STENDRA before nitrate administration is regarded as. Such circumstances, nitrates should still only be administered under close medical supervision with appropriate hemodynamic monitoring [see Contraindications ].
Connection between STENDRA on Blood pressure levels When Administered with Alpha-Blockers
1-center, randomized, double-blinded, placebo-controlled, two-period crossover trial was conducted to investigate the actual possibility interaction of STENDRA with alpha-blocker agents in healthy male subjects which contains two cohorts:
Cohort A (N=24): Subjects received oral doses of doxazosin once daily in the morning at 1 mg for 1 day (Day 1), 2 mg for just two days (Days 2 – 3), 4 mg for 4 days (Days 4 – 7), and 8 mg for 11 days (Days 8 – 18). On Days 15 and 18, the topics also received an individual oral dose of either 200 mg STENDRA or placebo, in line with the treatment randomization code. The STENDRA or placebo doses were administered 1.three hours following your doxazosin administration on Days 15 and 18. The co-administration function is to ensure doxazosin (Tmax ~a couple of hours) and STENDRA (Tmax ~0.7 hours) would reach their peak plasma concentrations at the same time.
Cohort B (N=24): Subjects received 0.4 mg daily oral doses of tamsulosin in the morning for 11 consecutive days (Days 1 – 11). On Days 8 and 11, the topics also received an individual oral dose of either 200 mg STENDRA or placebo, good treatment randomization code. The STENDRA or placebo doses were administered 3.3 hours following tamsulosin administration on Days 8 and 11. The co-administration principal purpose is so tamsulosin (Tmax ~4 hours) and STENDRA (Tmax ~0.7 hours) would reach their peak plasma concentrations together.
Supine and sitting BP and pulse measurements were recorded before and after STENDRA or placebo dosing.
An overall total of seven subjects in Cohort A (doxazosin) experienced potentially clinically important absolute values or changes from baseline in standing SBP or DBP. Three subjects experienced standing SBP values less than 85 mmHg. One subject experienced a decrease from baseline in standing SBP over 30 mmHg following STENDRA. Two subjects experienced standing DBP values a lot less than 45 mmHg following STENDRA. Four subjects experienced decreases from baseline in standing DBP over 20 mmHg following STENDRA. One subject experienced such decreases following placebo. There were no severe adverse events in connection with hypotension reported through the trial. There are no cases of syncope.
Earnings of five subjects in Cohort B (tamsulosin) experienced potentially clinically important absolute values or changes from baseline in standing SBP or DBP. Two subjects experienced standing SBP values a lot less than 85 mmHg following STENDRA. One subject experienced a decrease from baseline in standing SBP greater than 30 mmHg following STENDRA. Two subjects experienced standing DBP values less than 45 mmHg following STENDRA. Four subjects experienced decreases from baseline in standing DBP over 20 mmHg following STENDRA; one subject experienced such decreases following placebo. There were no severe adverse events associated with hypotension reported through the trial. There are no cases of syncope.
Table 5 presents the placebo-subtracted mean maximum decreases from baseline (95% CI) in systolic blood pressure latest results for the 24 subjects who received STENDRA 200 mg and matching placebo.
Table 5: Placebo-Subtracted Mean (95% CI) Maximum Decreases from Baseline in Standing and Supine Systolic Blood pressure levels (mmHg) with 200 mg STENDRA
|
Doxazosin Supine Standing |
-6.0 (-9.1, -2.9) -2.5 (-6.5, 1.5) |
|
Tamsulosin Supine Standing |
-3.1 (-6.4, 0.1) -3.6 (-8.1, 0.9) |
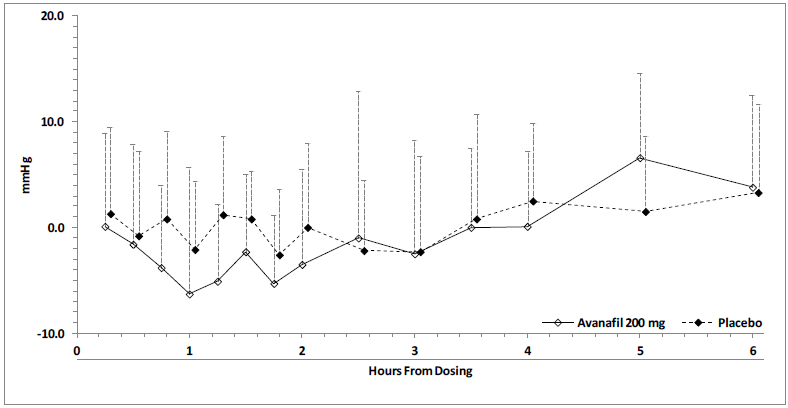
Hypertension effects (standing SBP) in normotensive men on stable dose doxazosin (8 mg) following administration of STENDRA 200 mg or placebo, are shown in Figure 2. Blood pressure effects (standing SBP) in normotensive men on stable dose tamsulosin (0.4 mg) following administration of STENDRA 200 mg or placebo are shown in Figure 3.
Figure 2: Mean (SD) Differ from Baseline in Standing Systolic Blood Pressure Over Time Following Administration of an Single Dose 200 mg Dose of STENDRA with Doxazosin
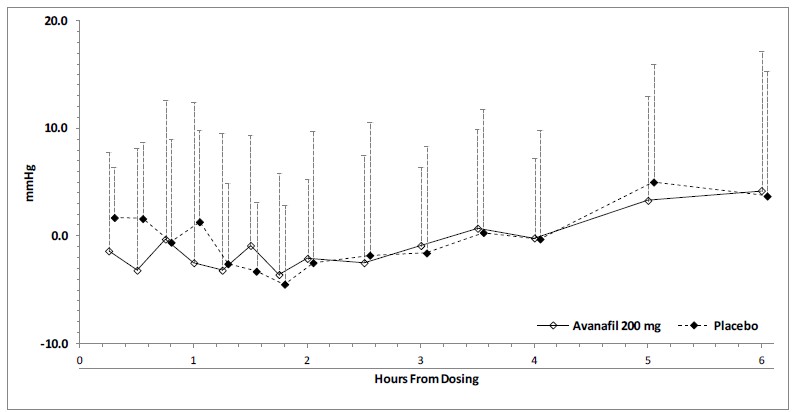
Figure 3: Mean (SD) Changes from Baseline in Standing Systolic Blood Pressure With time Following Administration of the Single Dose 200 mg Dose of STENDRA with Tamsulosin
Connection between STENDRA on Bp When Administered with Enalapril
An endeavor was conducted to evaluate the interaction of enalapril (20 mg daily) and STENDRA 200 mg. Single doses of 200 mg STENDRA co-administered with enalapril caused a mean maximum lessing of supine systolic/diastolic hypertension of a single.8/3.5 mmHg (compared to placebo), plus a mean maximum improvement in pulse of a single.0 bpm.
Upshots of STENDRA on Blood Pressure When Administered with Amlodipine
An endeavor was conducted to evaluate the interaction of amlodipine (5 mg daily) and STENDRA 200 mg. Single doses of 200 mg STENDRA co-administered with amlodipine caused a mean maximum decline in supine systolic blood pressure level of 1.2 mmHg (as compared to placebo), accompanied with a mean maximum rise in pulse rate of just one.0 bpm; the mean maximum decline in diastolic hypertension was below that noticed in the placebo group. There is no effect of STENDRA on amlodipine plasma concentrations. Concomitant amlodipine was related to 22% and 70% increases in avanafil Cmax and AUC, respectively.
Connection between STENDRA on Hypertension When Administered with Alcohol
Alcohol and PDE5 inhibitors, including STENDRA, are mild systemic
vasodilators. The interaction of STENDRA with alcohol was evaluated in a very
clinical pharmacology trial. Alcohol was administered in a dose of 0.5 g/kg,
which can be comparable to approximately
Effects of STENDRA on Semen
One particular 200 mg dose of STENDRA had no acute effect on sperm motility or sperm morphology inside of a number of healthy male subjects. The effect of avanafil on human spermatogenesis is unknown.
Upshots of STENDRA on Vision
Single oral doses of Type 5 phosphodiesterase inhibitors have demonstrated transient dose-related impairment of color discrimination (blue/green), utilizing the Farnsworth-Munsell 100-hue test, with peak effects nearby the time of peak plasma levels. This finding is similar to the inhibition of PDE6, which is involved with phototransduction inside the retina.
Pharmacokinetics
Mean STENDRA plasma concentrations measured following on from the administration of an individual oral dose of 50 or 200 mg to healthy male volunteers are depicted in Figure 4. The pharmacokinetics of STENDRA are dose proportional from 12.5 to 600 mg.
Figure 4 Plasma Avanafil Concentrations (mean ± SD) After a Single 50 mg or 200 mg STENDRA Dose
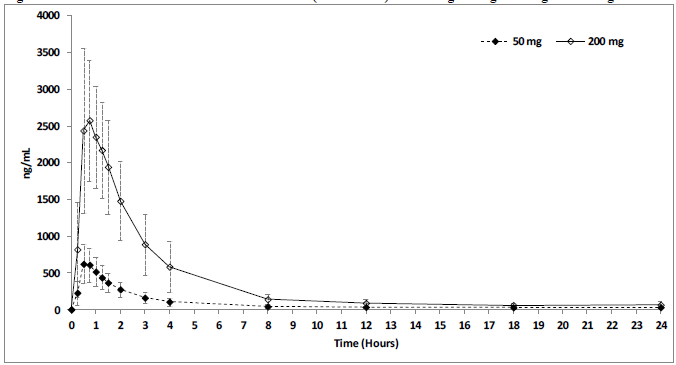
Absorption and Distribution
STENDRA is rapidly absorbed after oral administration, having a median Tmax of 30 to 45 minutes within the fasted state. When STENDRA (100 mg or 200 mg) is taken which has a high-fat meal, the rate of absorption is reduced, which has a mean delay in Tmax of 1.12 to a single.25 hours and a mean cut in Cmax of 24% (100 mg) and 39% (200 mg). Clearly there was approximately 3.8% loss of AUC. The tiny changes in avanafil Cmax and AUC are viewed as of minimal clinical significance; therefore, STENDRA may be administered with or without food. The mean accumulation ratio is around 1.2. Avanafil is approximately 99% likely to plasma proteins. Protein binding is independent of total drug concentrations, age, renal and hepatic function.
Avanafil wasn't found to build up in plasma when 200 mg doses of STENDRA were administered twice daily over 7 days.
In relation to measurements of avanafil in semen of healthy volunteers 45-90 minutes after dosing, below 0.0002% of your administered dose appeared from the semen of patients.
Metabolism and Excretion
Avanafil is cleared predominantly by hepatic metabolism, largely by the CYP3A4 enzyme as well as a small extent by CYP2C isoform. The plasma concentrations of your major circulating metabolites, M4 and M16, are approximately 23% and 29% that from the parent compound, respectively. The M4 metabolite carries with it an in vitro inhibitory potency for PDE5 18% of these of avanafil and M4 makes up about approximately 4% of your pharmacologic activity of avanafil. The M16 metabolite was inactive against PDE5.
Avanafil was extensively metabolized in humans. After oral administration, avanafil is excreted as metabolites predominantly in the feces (approximately 62% of administered oral dose) and to a lesser extent within the urine (approximately 21% in the administered oral dose). STENDRA contains a terminal elimination half-life of approximately 5 hours.
Geriatric
The pharmacokinetics on the single 200 mg STENDRA administered to fourteen healthy elderly male volunteers (65-80 years) and eighteen healthy younger male volunteers (18-43 years old) were compared. AUC0-inf increased by 6.8% and Cmax decreased by 2.1% in the elderly group, than the younger group. However, greater sensitivity to medications using some older individuals might be of interest [see Use within Specific Populations ].
Renal Impairment
The pharmacokinetics of an single 200 mg STENDRA administered to nine patients with mild (creatinine clearance above or of about 60 and less than 90 mL/min) and to ten patients with moderate (creatinine clearance greater than or add up to 30 to fewer than 60 mL/min) renal impairment were evaluated. AUC0-inf decreased by 2.9% and Cmax increased by 2.8% in patients with mild renal impairment, as compared to healthy volunteers with normal renal function. AUC0-inf increased by 9.1% and Cmax decreased by 2.8% in patients with moderate renal impairment, in comparison to healthy volunteers with normal renal function. There isn't any data readily available for subjects with severe renal insufficiency or end-stage renal disease on hemodialysis [see Use in Specific Populations ].
Hepatic Impairment
The pharmacokinetics of any single 200 mg STENDRA administered to eight patients with mild hepatic impairment (Child-Pugh A) and eight patients with moderate hepatic impairment (Child-Pugh B) were evaluated. AUC0-inf increased by 3.8% and Cmax decreased by 2.7% in patients with mild hepatic impairment, compared to healthy volunteers with normal hepatic function. AUC0-inf increased by 11.2% and Cmax decreased by 51% in patients with moderate hepatic impairment, in comparison to healthy volunteers with normal hepatic function. There's no data intended for subjects with severe hepatic impairment (Child-Pugh Class C) [see Utilization in Specific Populations ].
Drug Interactions
Effect of CYP3A4 Inhibitors on Avanafil: Strong and moderate CYP3A4 inhibitors increase plasma concentrations of STENDRA. The result of strong CYP3A4 inhibtors, ketoconazole and ritonavir, and moderate CYP3A4 inhibitor, erythromycin, on avanafil pharmacokinetics was studied in the open-label, randomized, one-sequence crossover, three-way parallel study.
Strong CYP3A4 Inhibitors
Fifteen healthy male volunteers received 400 mg ketoconazole (2 tablets containing 200 mg ketoconazole) once daily for 5 days (Days 2-6) and also a single 50 mg avanafil on Days 1 and 6. Twenty-four hour pharmacokinetics of avanafil on Days 1 and 6 were compared. Co-administration together with the strong CYP3A4 inhibitor ketoconazole generated an approximate 13-fold boost in AUC0-inf and 3.1-fold development of Cmax. Fourteen healthy male volunteers received 300 mg ritonavir (3 tablets containing 100 mg ritonavir) two times a day for 1 day (Day 2), 400 mg two times a day for 1 day (Day 3), 600 mg twice a day for 5 days (Days 4-8), as well as a single 50 mg avanafil on Days 1 and 8. Twenty-four hour pharmacokinetics of avanafil on Days 1 and 8 were compared. Co-administration together with the strong CYP3A4 inhibitor ritonavir resulted in an approximate 13-fold rise in AUC0-inf and 2.4-fold increase in Cmax of avanafil
Moderate CYP3A4 Inhibitors
Fifteen healthy male volunteers received 500 mg erythromycin (2 tablets containing 250 mg erythromycin) every 12 hrs for five days (Days 2-6) and a single 200 mg avanafil (2 tablets containing 100 mg avanafil) on Days 1 and 6. Twenty-four hour pharmacokinetics of avanafil on Days 1 and 6 were compared. Co-administration using the moderate CYP3A4 inhibitor erythromycin generated approximately 3.6-fold boost in AUC0-inf and a couple.0-fold rise in Cmax of avanafil.
Effect of Avanafil on Other Drugs:
Warfarin
The effects of avanafil on warfarin pharmacokinetics and pharmacodynamics was evaluated within a double-blind, randomized, placebo-controlled, two-way crossover study. Twenty-four healthy male volunteers were randomized to obtain either 200 mg avanafil or matching placebo for 9 days. On Day 3 of each and every period, volunteers received a particular 25 mg warfarin. Pharmacokinetics of R- and S-warfarin, PT, and INR ahead of warfarin dosing and up to 168 hrs after warfarin administration were compared. Platelet aggregation prior to warfarin dosing and assend to 24 hrs after warfarin administration were compared. PT, INR, and platelet aggregation could not change with avanafil adminstration: 23.1 sec, 2.2, and 75.5%, respectively. Co-administration with avanafil resulted in approximately 1.6% surge in AUC0-inf and 5.2% decrease in Cmax of S-warfarin.
Omeprazole, Rosiglitazone, and Desipramine
The result of avanafil around the pharmacokinetics of omeprazole (a CYP2C19 substrate), rosiglitazone (a CYP2C8 substrate), and desipramine (a CYP2D6 substrate) was evaluated within the open-label, three cohort, crossover study. Nineteen healthy male volunteers received a single 40 omeprazole delayed-release capsule once daily for 8 days (Days 1-8), including a single 200 mg avanafil on Day 8. Twelve hour pharmacokinetics of omeprazole on Days 7 and 8 were compared. Co-administration with avanafil ended in an approximate 5.9% increase in AUC0-inf and 8.6% increase in Cmax of omeprazole. Twenty healthy male volunteers received a single 8 mg rosiglitazone tablet then a single 200 mg avanafil. Twenty-four hour pharmacokinetics of rosiglitazone with and without avanafil were compared. Co-administration with avanafil resulted in an approximate 2.0% boost in AUC0-inf and 14% loss of Cmax of rosiglitazone. Twenty healthy male volunteers received a single 80 mg desipramine tablet a single 200 mg avanafil. Ninety-six hour pharmacokinetics of desipramine with and without avanafil were compared. Co-administration with avanafil lead to approximately 5.7% increase in AUC0-inf and 5.2% decrease in Cmax of rosiglitazone.
-
 Print This
Print This
-
 Share
Share